Beta-thalassemia er en arvelig blodsykdom forårsaket av genmutasjoner der kroppen lager for lite hemoglobin — proteinet i røde blodlegemer som fører oksygen gjennom kroppen. I denne tilstanden ødelegges også røde blodlegemer. Over tid kan utilstrekkelig hemoglobin føre til anemi (lavt antall røde blodlegemer eller RBC) og andre komplikasjoner. Personer med den alvorligste forstyrrelsesformen, beta-thalassemia major eller Cooley's anemi, blir vanligvis behandlet med regelmessige blodtransfusjoner; de med mindre type, beta-thalassemia intermedia, kan trenge transfusjoner sjeldnere.
Violka08 / Getty ImagesTyper
Det er tre typer beta-thalassemia.
- Beta-thalassemia-egenskap, der en person bærer genet for sykdommen, men ikke har symptomer. Noen ganger kalles det thalassemia minor.
- Beta-thalassemia intermedia, en relativt mild form for lidelsen
- Beta-thalassemia major, eller Cooley’s anemia, som er den mest alvorlige formen
Beta-thalassemia er mest utbredt blant mennesker av middelhavs, asiatisk, indisk og afrikansk avstamning.
Symptomer
Symptomene på beta-thalassemia avhenger av typen og alvorlighetsgraden av lidelsen. Personer med beta-thalassemia minor kan ha veldig mild anemi, men utvikler vanligvis ikke merkbare symptomer. Noen vet kanskje ikke engang at de bærer det forandrede genet.
Selv om de to muterte HBB-gener er tilstede ved fødselen, kan babyer født med enten beta-thalassemia intermedia eller Cooley's anemi ikke ha symptomer før mellom 3 og 6 måneder; noen barn med beta-thalassemia utvikler ikke symptomer før de er 2 år.
Uansett når et barn med beta-thalassemi blir symptomatisk, vil de sannsynligvis oppleve følgende:
- Anemi
- Utmattelse
- Svakhet
- Kortpustethet
- Gulaktig eller blek hud
- Langsom vekst
- Mørk urin
- Liten appetitt
- Oppstyr
For de med Cooley's anemi kan symptomene utvikle seg og komplikasjoner kan utvikles, for eksempel:
- Hevelse i magen
- Bendeformasjoner eller knuste bein på grunn av endringer i benmargen der det produseres røde blodlegemer
- Splenomegali, en tilstand der milten, som filtrerer røde blodlegemer, er overarbeidet og forstørres
- Infeksjoner
Diagnose
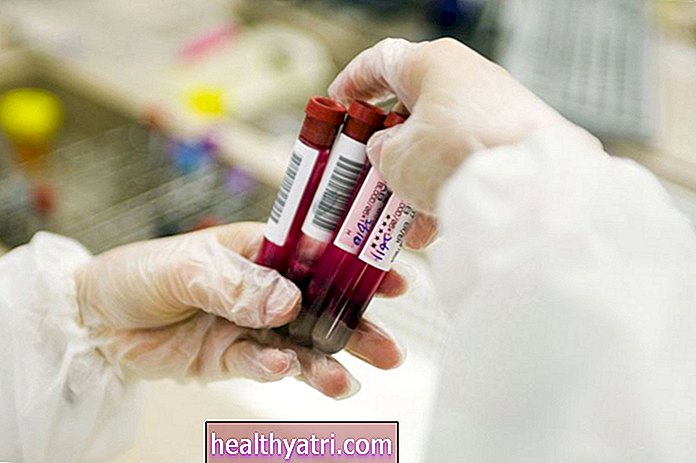
Selv om thalassemia major vil bli identifisert på den nyfødte skjermen, kan det hende at personer med thalassemia intermedia ikke blir identifisert før år senere. Disse menneskene blir vanligvis identifisert ved rutinemessig fullstendig blodtelling (CBC). CBC vil avdekke en mild til moderat anemi med svært små røde blodlegemer. Dette kan forveksles med jernmangelanemi.
Diagnosen er bekreftet av en hemoglobinprofil (også kalt elektroforese). I beta-thalassemia intermedia og trekk avslører denne testen forhøyelse av hemoglobin A2 (en andre form for voksen hemoglobin) og noen ganger F (foster). Alfa-thalassemia intermedia kalles vanligvis hemoglobin H-sykdom, da dette er det dominerende hemoglobinet som ses på profilen.
Beta-thalassemia kan diagnostiseres gjennom forskjellige blodprøver. Moderat og alvorlig beta-thalassemi blir vanligvis diagnostisert i tidlig barndom eller når du er 2 år gammel fordi tegn og symptomer som anemi vanligvis forekommer veldig tidlig. Hvis foreldrene dine har sykdommen, kan du ha blitt testet og diagnostisert ved fødselen eller som småbarn, spesielt hvis du utviste anemi i veldig ung alder.
De forskjellige testene som brukes til å diagnostisere beta-thalassemia inkluderer:
- Den nyfødte skjermen, som testes rutinemessig til de fleste spedbarn ved fødselen for å identifisere eventuelle arvelige forhold. Dette kan oppdage beta-thalassemia eller, i det minste, rød-flagg karakteristikk av sykdommen i blodet for videre overvåking av en hematolog. Den nyfødte skjermen innebærer å ta en liten blodprøve og sende den til et laboratorium for evaluering.
- En komplett blodtelling (CBC), som måler hemoglobin og mengden og størrelsen på røde blodlegemer. CBC vil også avsløre en mild til moderat anemi med svært små røde blodlegemer, som er typiske for beta-thalassemi. (Noen ganger kan imidlertid en diagnose forveksles med jernmangelanemi)
Personer som har mildere former for talassemi kan bli diagnostisert etter at en rutinemessig blodprøve viser at de har anemi. Leger kan også mistenke og teste for beta-thalassemia hos mennesker av afrikansk, middelhavs eller øst-asiatisk avstamning som ser ut til å ha anemi.
- Et antall retikulocytter, som bestemmer helsen til benmarg og ofte er en del av en opparbeidelse når man tester for anemi, kan indikere at beinmarg ikke produserer et tilstrekkelig antall røde blodlegemer.
- Kontroll av jernnivået i blodet vil vise om årsaken til anemi er jernmangel eller talassemi.
Siden beta-thalassemia er en genetisk lidelse som overføres fra foreldre til barn, flere andre tester som kan bidra til å forutsi tilstedeværelsen av genet, og mulig alvorlighetsgrad av beta-thalassemia:
- Hemoglobinelektroforese, som er en blodprøve for foreldre designet for å oppdage forskjellige genetiske abnormiteter i hemoglobinstrukturen, kan vise nivåene av hemoglobin.
- Prenatal testing kan avsløre alvorlighetsgraden av beta-thalassemia i tilfeller der foreldrene bærer genet. Disse kan omfatte: prøvetaking av korionvillus utført rundt den 11. uken av svangerskapet, og fjernet en liten bit av morkaken for evaluering; og fostervannsprøve, som gjøres rundt den 16. uken av svangerskapet og innebærer å undersøke en prøve av væsken som omgir fosteret.
Behandling
Standardbehandlingene for personer med moderat eller alvorlig beta-thalassemi er blodtransfusjoner, jernchelasjonsbehandling og folinsyretilskudd:
- Blodtransfusjoner: En transfusjon av røde blodlegemer kan kompensere for mangelen og sørger for normale hemoglobinnivåer. Mennesker med betatestalassemi major trenger ofte transfusjoner annenhver eller fjerde uke gjennom hele livet; de med beta-thalassemia intermedia kan bare kreve sporadiske transfusjoner i perioder med sykdom eller pubertet.
- Jerncheleringsterapi: Dette hjelper kroppen å fjerne overflødige og potensielt skadelige mengder jern fra blodet - som kan skyldes mange transfusjoner. Jernchelering administreres oralt eller gjennom en infusjon under huden.
- Splenektomi: Bruk av denne prosedyren har avvist de siste årene, men fjerning av milten blir noen ganger vurdert når kelasjonsbehandling ikke fungerer.
- Folinsyretilskudd: Folsyre er et B-vitamin som hjelper til med å bygge sunne røde blodlegemer. Legen din kan anbefale folinsyretilskudd i tillegg til blodtransfusjoner og jernchelasjonsbehandling.
- Benmarg og stamcelletransplantasjon: En blod- og margtransplantasjon erstatter defekte stamceller med sunne fra en annen person (en donor). Stamceller er cellene i benmargen som lager røde blodlegemer. Selv om dette kan være en mulig kur mot beta-thalassemia, er det bare et lite antall mennesker som har alvorlig thalassemia som kan finne en god donormatch for denne risikable prosedyren.
Mennesker som er beta-thalassemia-bærere eller som har blitt identifisert med egenskapen, men som har milde symptomer eller ingen i det hele tatt, krever vanligvis veldig lite ved behandling.
Et ord fra veldig bra
Hvis du eller familiemedlemmer har beta-thalassemia eller genet for beta-thalassemia, er det lurt å snakke med legen din hvis du tenker på å få barn. En genetisk rådgiver kan bidra til å bestemme risikoen for å overføre lidelsen til avkommet ditt. Hvis du forventer en baby og du og partneren din er thalassemia-bærere, kan det være lurt å vurdere prenatal testing. Selv om disse testene riktignok virker skumle og forstyrrende, vil de faktisk lindre angsten din og hjelpe deg å være forberedt, uansett utfall.



-count.jpg)













-is-treated.jpg)